
Recent Zimmer Hips/Shoulder/Knee News & Information
Zimmer Biomet has agreed to pay $17.3 million in criminal penalties, as well as a civil settlement of $5.4 million to the U.S. Securities and Exchange Commission.
In February of 2016, Zimmer Biomet agreed to pay a settlement of $350,000 to Colorado man.
Zimmer Biomet is the second-largest medical device manufacturer in the world, each year selling more than 1 million artificial knee, shoulder, hip, ankle, foot and elbow implants. The company has over $4.5 billion in annual sales.
Zimmer Biomet Holdings, an Indiana-based company, is one of the leading manufacturers of joint replacement and orthopedic products. Zimmer Biomet sells more than 1 million artificial hip, shoulder, and knee replacement implants annually. The acquisition of Biomet Orthopedics in 2014 made Zimmer the second-largest manufacturer in the orthopedic market. Its annual revenue for 2018 was $7.9 billion. While most of the products manufactured by this company have been successful, several devices have resulted in mass tort actions and recall over the last several years. Thousands of people have taken legal action against the manufacturer alleging high failure rates, revision surgeries, and serious injuries.
If you suffered complications after receiving a Zimmer Biomet hip, shoulder or knee implant during your operation, the lawsuit experts at Consumer Alert Now would like to speak with you. By joining a Zimmer Biomet mass tort, you may be entitled to compensation for injuries or the loss of a loved one. Contact us at 800-511-0747 for a free case evaluation.
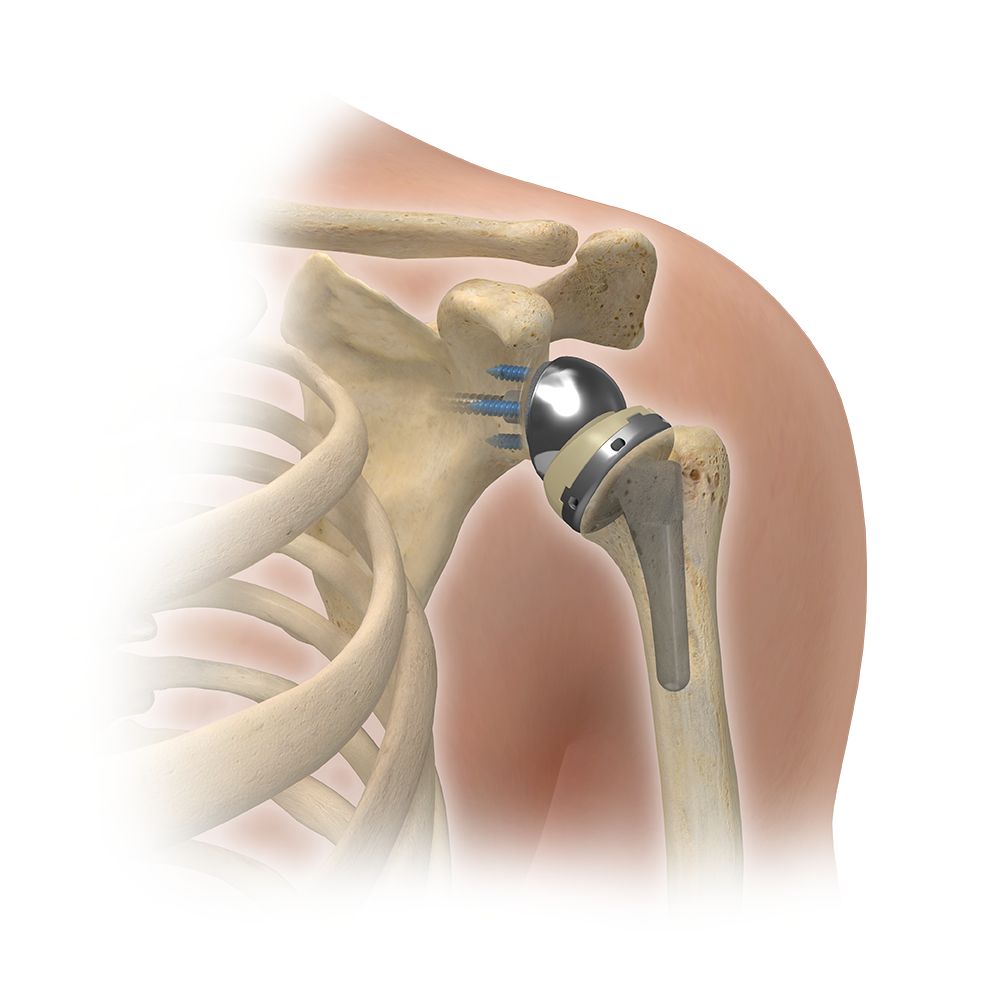
Zimmer Biomet Comprehensive Reverse Shoulder Device
Biomet Inc., which has since been acquired by Zimmer Holdings, launched its Comprehensive Reverse Shoulder System Humeral Tray in 2008. This is a complex medical device designed to replace the shoulder joint and restore a patient’s arm movements. The device was produced from August 2008 to September 2012. The device was approved by the Food and Drug Administration (FDA) through the controversial 510(k) Fast-Track Program, which allows manufacturers to show that their new device is “substantially similar” to another device already being sold and distributed. Biomet was able to show that the device was similar to shoulder replacement devices that had already been approved by the FDA. This means that the device was made available in the medical market, having bypassed the need for human clinical trials.
The Purpose of a Reverse Shoulder Implant
A conventional shoulder replacement implant mimics the human shoulder anatomy. The shoulder is a socket and ball joint, where the humerus (upper arm bone) has a ball-shaped end that fits into a socket on the shoulder blade. With a conventional shoulder implant, the natural ball on the humerus is removed and replaced with a metal ball. A stem is placed into the center of the arm bone and connected to artificial bone. Conversely, the natural socket is replaced with a plastic cup, and the metal ball is snapped into place.
Conventional shoulder replacement implants do not work for all patients, especially those suffering from cuff tear arthropathy (damaged rotator cuffs). Rotator cuffs are muscles and tendons that hold the ball into the socket in a natural shoulder joint. A conventional shoulder replacement implant relies on rotator cuffs to move the arm. A reverse shoulder replacement implant is designed in a manner that reverses the shoulder’s natural anatomy.
The reverse shoulder may be a better option for a patient who has a torn or damaged rotator cuff, has had a previous, failed shoulder replacement, or has extreme weakness or inability to lift the arm beyond 90 degrees. With a reverse shoulder replacement, the location of the ball and the socket are reversed. The socket is attached to the arm bone while the ball is attached to the shoulder blade. This procedure allows patients to lift the arm using the deltoid muscles instead of the rotator cuff.
Comprehensive Reverse Shoulder Device Complications
Patients who were implanted with a Zimmer Biomet Comprehensive Reverse Shoulder device from October 2008 to September 2015 are at great risk for injury. According to reports, the complication rate of reverse shoulder replacement devices is as high as four times that of conventional shoulder-replacement devices. A 2013 study involving nearly 3,000 reverse shoulder-replacement patients found that the need for revision surgery in patients younger than 60 was at twice the rate of those 60 years or older.
Reported complications of the Zimmer Biomet Comprehensive reverse-shoulder replacement implants, include:
- High fracture rate
- Component loosening or failure
- Dislocation of the ball and socket of the implant
- An infection which may require surgery to treat
- Bone fractures in the shoulder blade or around the implant
- Weakness, tingling sensation or numbness
- Scapular notching- wearing a groove into the shoulder blade
- Hematomas-pockets of blood-forming in the joint area
The Zimmer reverse shoulder has the potential risk of causing the patient severe pain, permanent loss of shoulder use, need for revision surgery, or death.
FDA Class I Recall
On December 15, 2016, Zimmer Biomet issued a voluntary recall of 3,662 Comprehensive Reverse Shoulder Humeral Implants that had been manufactured between August 2008 and September 2011 and distributed between October 2008 and September 2015. The manufacturer recalled the devices because the fracture rate was higher than what was indicated in the labeling. The FDA identified this as a Class 1 recall, which is the agency’s most serious type of recall issued when a product carries the risk of causing serious injuries or death. In their attempt to resolve problems caused by the reverse shoulder implant, doctors have had to recourse to a post-operational surgical correction, which according to the FDA, is an invasive process that could cause severe health complications, including infection, permanent loss of shoulder functions, or death.
Based on reports from Zimmer’s recalls, the problem appears to be caused by a malfunction in the device’s “humeral tray.” This refers to a composing piece that has a tendency to break away from the device. According to the manufacturer, their product has a consumer warning label, but the FDA stated that the fracture rate was higher than what was originally stated by the manufacturer.
This is not the first time that Zimmer Biomet’s Reverse Shoulder Systems have been recalled. In 2015, the FDA issued a Class II Recall of the Comprehensive Reverse Shoulder Glenosphere Mini Baseplate with Taper Adaptor. In 2010, the manufacturer recalled 47 units of the Comprehensive Reverse Shoulder Humeral Tray with Locking Ring due to reports of the device fracturing.
Zimmer Biomet Reverse Shoulder Mass Tort
Victims of defective reverse shoulder replacement implants manufactured by Zimmer Biomet have filed a mass tort action against Zimmer Biomet. As of June 2019, judges have not consolidated the many lawsuits filed in court into a Multidistrict Litigation (MDL). However, with such a big number of mass torts and personal injury lawsuits filed in state and federal courts, it’s just a matter of time before centralization occurs to allow for mass tort litigation.
The only publicly reported settlement involving Zimmer Comprehensive Reverse Shoulder System Humeral Tray was filed in 2014 by a Colorado man. In his complaint, he claimed that the manufacturer had received multiple reports of complications associated with the device, but continued to sell “defective and unreasonably dangerous devices.” In 2016, Zimmer Biomet agreed to settle the litigation with a $350,000 cash settlement.
Zimmer Hip Replacement
Zimmer Biomet is the largest hip replacement product manufacturer in the United States. Zimmer hip replacement devices involve a series of artificial joint and parts that primarily use metal-on-metal technology. Hip joint function is based on the design of a ball and a socket. A ball-shaped protrusion on top of the leg fits into a socket in the hip to allow for free movement of the leg. The ball-and-cup system can be replaced with a hip replacement device when the structure and connection of the hip breaks. In theory, this technology should ensure a stronger and more flexible hip. Also, it was intended to prolong the lifespan of a hip replacement device as they are supposedly more durable and can last longer. These forms of hip replacement products have been heavily marketed as ideal for younger patients and those who intended to remain active.
However, this metal-on-metal construction has been blamed for Zimmer hip replacement side effects. The first issue with Zimmer hip replacements included the Durom Acetabular Component, commonly known as the Durom Cup. Mass tort actions followed shortly after the device arrived on the market in 2006. Patients who had received the device claimed that it was failing prematurely and caused serious complications requiring revision surgery to correct the problem. The device was designed to fuse to the hip socket without the use of screws or cement but failed to fuse I some patients. Zimmer temporarily recalled the device in 2008, but doctors had already implanted the device in about 12,000 patients. Zimmer Biomet said the recall was meant to allow the company to issue new implantation instructions. The FDA said that the recall was based on false and misleading labeling.
The Durom Cup was returned to the market with new instructions on October 2009, but the company discontinued the product in 2010. That same year, all Durom Cup lawsuits were consolidated into multidistrict litigation (MDL). The company had faced more than 700 lawsuits in the New Jersey federal court. Zimmer set up a $47.5 million settlement fund for the mass torts. The company spent several years settling lawsuits before they went to trial. In March 2016, Zimmer offered to settle a majority of the Durom Cup lawsuits in the MDL for $314 million. A large number of plaintiffs involved in the mass tort were no content with the settlement offer and chose to reject it. As of May 2019, there were 97 cases pending in the Durom Cup New Jersey MDL.
Zimmer Versys Femoral Head, M/L Taper Hip Prothesis, and Kinectiv Technology
In October 2018, all other Zimmer hip implants lawsuits were consolidated into a new MDL. The mass tort action targeted Zimmer’s Versys Hemoral Head, M/L Taper Hip Prosthesis, as well as versions using Kinectiv Technology. The M/L Taper Hip Prostheses were made from a titanium alloy using the metal-on-metal design. The Versys Femoral Head is manufactured from a cobalt-chromium alloy.
The mass tort action alleges that Zimmer VerSys Hip femoral head and Zimmer M/L Taper Hip Prosthesis are patently defective and dangerously incompatible when implanted together, with a tendency to corrode between the components. The wear affected the junction where the neck and head of the implant are joined, causing trunnionosis. This can generate toxic metallic debris into the bloodstream and the nearby tissues, leading to metallosis, pseudotumors, the body rejecting the hip replacement, and other serious complications. Consequently, the premature failure of the implant necessitates risky removal surgery.
In August 2006, The FDA issued a Class II recall of the VerSys Hip System for lack of assurance of proper metal fatigue strength due to the anomaly of the metal grain structure. In April 2015, Zimmer’s M/L Taper with Kinectiv Technology was removed from the market after being on the market for less than a month. The FDA later issued an urgent Class 1 recall of 752 implants due higher than allowed cytotoxicity levels found with the product.
The first 21 lawsuits were sent to the United States District Court for the Southern District of New York before Judge Paul Crotty. There were 59 cases pending in the MDL as of April 2019.
Adverse events that may be caused by Zimmer hip replacement products include:
- Metallosis- Tissue toxicity resulting from metal corrosion and flaking of microscopic or some larger pieces that are deposited into body tissues.
- Osteonecrosis- tissue death in areas around the implant caused by toxicity resulting from metal fragments. When tissues die, oxygen in the blood cannot be supplied to bone tissue, a condition that may cause the dissolution of the bone itself. Additional reconstructive surgery may be required since bone tissue will likely not grow back.
- Periprosthetic fractures- fractures of the hip implant which may require surgery to correct or remove the device
- Heterotopic ossification- bone tissue that develops outside to the skeleton into the soft tissues. This is extremely painful and can cause permanent damage.
- Pseudotumors- these are large collections of mass surrounding the implant, which cause severe pain, inflammation, and restrict movement.
Zimmer Biomet Knee Replacement Devices
Knee replacements are marketed as effective methods of increasing mobility and reducing pain in patients. However, knee replacement can cause severe complications for some patients, especially when the device is defective. In recent years, thousands of knee replacement lawsuits have been filed with most alleging that the devices loosened due design defect or other flaws.
There are various types of knee replacements. Zimmer Biomet, the largest-knee implant maker in the world, makes devices for both total and partial knee replacements. There are variations intended to accommodate patients based on their needs, gender, activity level, age, and other considerations. Also, the knee replacement devices comprise of many different components, including plates, screws, and stems. Knee replacement devices are generally expected to last for 15-20 years. However, patients who received Zimmer knee replacement devices experience extreme pain and early failures of their devices, often requiring revision surgery to correct or replace the knee implant. Other Zimmer knee complications include ascetic loosening off the joints, difficulty walking or bending of the knee, and radiolucent line on examination of the knee.
Zimmer Biomet Knee Replacement Recalls
Since 2003, Zimmer has issued at least 355 recalls related to their knee replacement devices. The most recent was issued in 2017 and affected more than 28,000 joint components. Inspections found that the parts had elevated levels of endotoxin.
In March 2017, a Class II recall was issued for 8,154 units of Regenerex 3 Peg Series A Patella Kneecap components. Zimmer made the recall due to the fact the pegs can chip off, filling the knee joint with metal debris and causing catastrophic failure. In March 2015, the Personal Trabecular Metal Tibial Plate was recalled due to reports of radiolucent lines gaps existing between pieces of the device and the bone as well as the loosening of the implant. This was a Class II recall that affected a total of 11,658 devices.
The FDA issued a Class II recall for Zimmer’s Segmental System. The agency stated that the system’s instructions did not indicate conditions that would cause excessive loading on the system’s polyethylene insert, which would cause device failure. This recall affected 5,305 units. Zimmer recalled more than 41,000 parts of its NexGen Complete Knee Solution MIS Stemmed Tibial Component in July 2014. The manufacturer stated the reason for the recall as the potential that the threads in the drop-down stem plug or extension were out of specification and faulty.
In 2010, Zimmer recalled 68,284 units of its NexGen Complete Knee Solution MIS Total Knee Stemmed Tibial Component due to complaints of loosening and need for revision surgery. The manufacturer warned doctors to ensure the device was fully cemented and use a drop-down stem with the tibial plate.
In essence, most Zimmer knee replacements recall were due to:
- Improper fit- implants could suffer damage when put component is forced into place
- Defective design- implants and tools were predisposed to fracture due to flawed design
- Early wear- parts wore out earlier than expected
- Loosening- since knee replacements loosen forcing patients to undergo revision surgery
- Packaging errors- faulty packages jeopardized the sterility of surgical tools
- Severe pain in the knee
- Nerve and blood vessel damage
Zimmer Knee Settlements
Currently, the only active Knee Implant Multidistrict litigation is the NexGen Knee Implant MDL. The mass torts and individual lawsuits were consolidated in August 2011 in the Northern District of Illinois before Judge Rebecca Pallmeyer. More than 17,000 lawsuits had been included in the MDL, but most were dismissed. On February 12, 2018, the judge announced a settlement in the remaining cases and ordered a stay of litigation. The exact amount of settlement remains unknown since Zimmer, and the plaintiffs agreed to keep the amount confidential.
FDA’s 510(k) Approval Process - No Safety Studies
Many of the faulty and recalled Zimmer orthopedic devices were fast-tracked through the FDA’s 510(k) approval process. This process allows medical device manufacturers to introduce a product to market without being subject to testing before being used on patients. When the 510(k) process was initiated in 1975, it was meant to protect the consumer by allowing “similar” devices to market without forcing the manufacturer to undergo expensive and time-consuming research, testing, and clinical trials which had already been done on the device used as the “predicate.” The FDA clears the device if the manufacturer is able to show that the device is substantially similar to a legally marketed device. The law allows the agency to request study data from a 510(k) applicant, but that step is rarely taken.
The FDA will determine the safety of a device if it has the same use and technological characteristics as the predicate device. Alternatively, the device has the same intended use but different technological characteristics as the predicate device. What’s important is for the manufacturer to show that the device is at least as effective and safe as the already approved device. In other words, the FDA is not examining if the device is effective or safe; it just agrees with the claim made by the manufacturer that the device is similar to an already approved device.
Many of the devices approved through the 510(k) process are highly likely to be subject to a recall. In fact, researchers found that such devices are 11.5 times more likely to be pulled out of the market than devices that were put through the more rigorous Pre-Market Approval process. This may explain why Zimmer orthopedic devices approved through the 510(k) experienced high rates of failure.
Zimmer Biomet Bribes to Secure Sales
In 2012 Zimmer Biomet settled with the U.S Department of Justice over allegations that the company had bribed state-employed healthcare providers in China, Brazil, and Argentina for help in winning business for its hip, shoulder, and knee devices. In December 2016, a revised settlement was announced after the company admitted additional misconduct in Brazil and Mexico in 2015. Investigators found that Zimmer Biomet paid briberies to foreign officials and masked the payments as scientific incentives, royalties, commissions, and consulting fees. The company paid $17.3 million in criminal penalties and $5.4 million in a civil settlement. The medical device manufacturer also agreed to maintain a compliance program.
Find a Zimmer Hip/Shoulder/Knee Mass Tort Attorney Near Me
If you have suffered significant damages from a faulty Zimmer hip, shoulder, or knee implant, it’s time to find a team of experts who can help you overcome an unexpected medical problem. The on-staff legal consultants at Consumer Alert Now are available to listen to your concerns and answer your questions. We help people all around the country get help from a mass tort attorney. Contact us today at 800-511-0747 or for a free case evaluation.










